Message from the Provost
March 25, 2020
Dear Colleagues:
I am deeply appreciative of the ongoing commitment of our Fairfield University faculty during these difficult circumstances. The context of the rapidly evolving circumstances related to Covid-19, and Fairfield University’s focus on social distancing and the health and well-being of our community also creates certain challenges for the research community.
The Fairfield University Institutional Research Board (IRB) for human subjects research, in consultation with the Associate Vice Provost for Scholarly, Creative, and Community Engagement, has issued revised standards related to human subjects-related visits during this time period.
Detailed information can be found on the IRB website: http://faculty.fairfield.edu/IRB/
In short, studies involving face-to-face interaction with participants with no direct therapeutic benefit should be postponed until further notice. All research involving direct contact with human subjects research visits may be performed remotely (e.g., by phone or Zoom) whenever possible. This may require submitting an amendment to the IRB for approval, the details for which can be found on the IRB website.
The IRB remains operational, although approvals may take longer than usual due to our current circumstances. All research that qualifies for exempt, expedited, QI, or delegation to an external IRB will continue to be processed year-round. Please contact IRB Chair Alison Kris at akris@fairfield.edu and continue to check the IRB website for updates: http://faculty.fairfield.edu/IRB/.
The Fairfield University community is deeply grateful for the continued research of our faculty and the dedication of the IRB.
— Jocelyn M. Boryczka, PhD
Associate Vice Provost for Scholarly, Creative, and Community Engagement
Professor of Politics
Can I continue my human subjects research?
See the below chart:
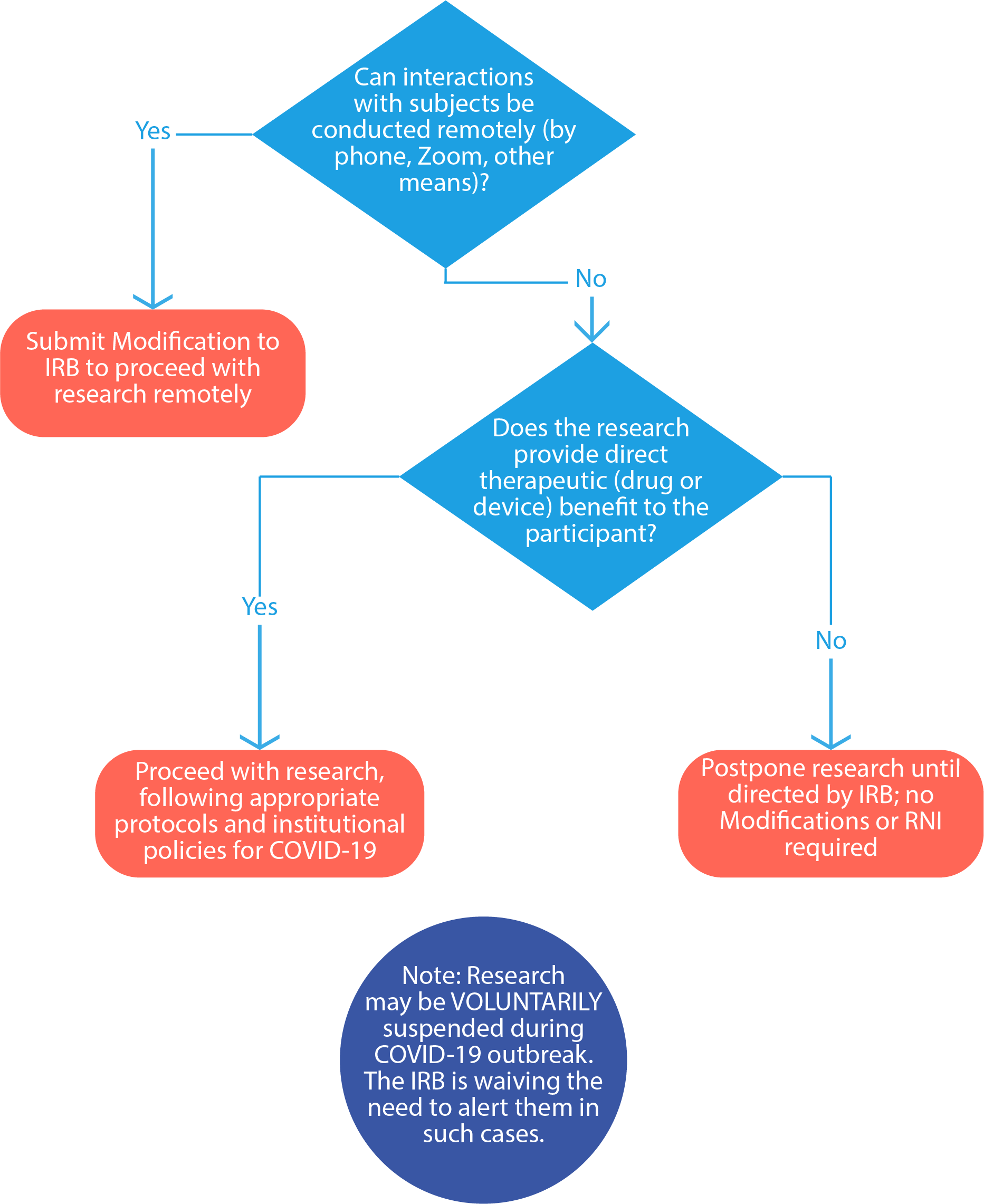
How can a PI modify study procedures to alter in-person visits to virtual/remote visits?
All modifications to research procedures require IRB review and approval prior to implementation, except those that are necessary to eliminate an immediate apparent hazard to a participant (these are then required to be submitted to the IRB afterward as an RNI [reportable new information]). If it is in the best interest of the subject to eliminate an immediate apparent hazard to one or more participants and there is no time to obtain prior IRB approval, a researcher may do so. The PI must then submit this modification as reportable new information (RNI) within 5 business days to the IRB for review.
If the PI would like to modify their procedures to replace in-person study visits with virtual/remote or phone options for administering procedures such as questionnaires, surveys, check-ins, screening, and consenting, these changes must be approved in advance by the IRB as a modification to the approved study. The PI should submit an amendment to their existing protocol (open the protocol, click submit sub-form, and select amendment).
Guidance for amendments to change to virtual/remote interactions with research participants:
For studies designated as Exempt that are conducted via phone or Zoom, the researcher might use a consent script he or she reads aloud that contains the relevant basic elements of consent (e.g., stating this is a research study, how long it will take, that subjects can discontinue their participation at any time, that participation is voluntary, and how data confidentiality will be maintained).
For studies designated as Exempt that involve online surveys, a researcher might use a written consent form that participants read online, and they indicate their consent by pressing a button, rather than by providing their signature. State that subjects indicate their consent by pressing a button agreeing to participate in the study. We recommend the researcher use the Consent Template for Online Research (DOC) | (PDF).
For studies designated as Expedited of Full Board review that involve online consent and collection of data, having participants press a button rather than sign their name technically is an "alteration of documentation of informed consent" since it is not a signed document. It is not a "waiver of informed consent" (waiver of informed consent = subjects do not have to agree to take part in the study). The researcher should review the information about requesting an alteration of documentation of informed consent in the section "Additional Information" below. We recommend that the researcher use the Consent Template for Online Research (DOC) | (PDF). They can create their own, but it must contain all the federally mandated required elements of consent: Section 116: General Requirements for Informed Consent Standards. The researcher does not need the traditional section headings for an online consent; however, the researcher does need to include the necessary information.
Can the PI decide to voluntarily pause, delay, or reschedule interactions with subjects until the COVID-19 outbreak has resolved?
Yes, the PI may decide voluntarily pause, delay, or reschedule interactions with subjects because of COVID-19; in such cases, there is no need to inform the IRB, regardless of review level. This is a temporary change to the IRB’s policy regarding submission of an RNI in these situations specifically related to the impact of COVID-19. However, you must:
- Ensure the voluntarily pause does not increase any risk to study subjects if your study is a greater-than-minimal risk study that involves some type of patient care.
- Contact study subjects who will be affected by this suspension (e.g., a study visit will be cancelled, etc.).
- Notify the study sponsor.
- Document the voluntary pause in the study records along with the justification and any actions taken, in the event that this is needed during an audit.
Student Research
Some professors will be converting their students’ research into something that is actually do-able in the current social-distancing environment. IRBs must review the types of research that are "systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge."
Some student projects are likely to be for the sake of learning within the class but are not designed or intended to contribute to generalizable knowledge. If they are not designed to contribute to generalizable knowledge, they do not fall under IRB purview.
For instance, a student may do an online survey that is a learning exercise and is not systematic research designed to contribute to generalizable knowledge. In that case IRB submission is not needed and the responsibility falls on the professor to ensure that reasonable research ethics are followed (e.g., protecting confidentiality, ensuring participation is voluntary). If it is a study designed to contribute to generalizable knowledge, then resubmit to the IRB and include a note to the Chair describing the intended timeframe.
Another solution is to have students create the materials as if they were going to run the study, but then not run it and create a fictional data set and report their fictionalized analyses. It is not as satisfying as real data but it does promote significant student learning.
There is some helpful information on Academic Twitter about converting student projects.
For Studies That Require Full Board Review
The IRB generally has its final meeting of the academic year in early May. Protocols for research that meet the criteria for exempt status, expedited review, QI, or delegation to an external IRB will continue to be processed by the IRB throughout the summer and can thus be submitted anytime during the summer. This is the vast majority of research that most of you do.
However, any researchers planning research over the summer that requires full board review -- for instance, research that involves more than minimal risk or that involves vulnerable populations — should to be submitted in advance of this final meeting in order to be reviewed.
Detailed information regarding definitions of "more than minimal
risk, "vulnerable populations," and what qualifies for full board
versus expedited or exempt research review can be found on the IRB
information page:
http://faculty.fairfield.edu/IRB/
When does a PI need to update their Clinicaltrials.gov registration?
Studies registered at the federal site ClinicalTrials.gov that are modifying their research procedures to include testing for SARS-CoV-2 and/or assessment of COVID-19 symptoms should update the ClinicalTrials.gov information to include these new procedures, if they are done for research purposes. If new procedures are being added as public health surveillance activities in coordination with public health authorities, the registration information does not need to be modified. Any research-related changes that are communicated to the subjects (past, ongoing, future) must be added to the study’s ClinicalTrials.gov registration with 30 days after IRB approval of the modification. See more guidance for NIH-funded clinical trials
We have adopted some of these materials and exact wording from Penn State University IRB’s website https://www.research.psu.edu/covid_irb.